
Computer System Validation Services: The Strategic Guide to GAMP 5 and GxP Compliance in 2026
- Jun 28
- 11 min read
Updated: Jul 5
Over 60% of regulatory findings in pharmaceutical inspections now stem from data integrity and computerized system failures. With FDA Warning Letters surging by 73% in 2025, the pressure to maintain airtight compliance has never been higher. You're likely buried under a mountain of validation documentation, fearing that a single manual error could trigger a Health Canada non-compliance report or a 483 observation. It's frustrating when critical project timelines stall because of outdated, slow-moving validation processes that feel more like a hurdle than a safeguard.
At APS Compliance Consultants Inc., we believe compliance should empower your growth, not hinder it. By mastering the strategic shift toward Computer Software Assurance (CSA) and the latest GAMP 5 standards, you can accelerate your project completion by 40% while ensuring your systems remain permanently audit-ready. This guide explores how to leverage expert computer system validation services to integrate 21 CFR Part 11 requirements into your daily workflows. We'll show you how to transform these complex regulatory burdens into a streamlined, high-performance competitive advantage for 2026 and beyond.
Table of Contents
Navigating the Regulatory Landscape of Computer System Validation (CSV) Services
Implementing a Risk-Based GAMP 5 Framework for GxP Compliance
The Modern Shift: Transitioning from Traditional CSV to Computer Software Assurance (CSA)
Streamlining the Validation Lifecycle: From Gap Analysis to Audit Readiness
Maximising ROI with Expert Validation Consulting and Integrated Software Solutions
Navigating the Regulatory Landscape of Computer System Validation (CSV) Services
Computer system validation services represent more than a technical hurdle; they're the essential documented evidence that your systems perform their intended functions consistently and reliably. In the current 2026 regulatory environment, "standard" testing protocols are no longer sufficient to satisfy inspectors. With the FDA issuing over 327 Warning Letters in just the first half of 2025, a 73% increase over the previous year, the margin for error has vanished. Modern compliance demands a proactive, risk-based approach that bridges the gap between global standards while maintaining the absolute integrity of GxP processes.
At its core, validation ensures that every piece of software and hardware used in GMP, GLP, or GCP environments operates within defined parameters. This is not a static event but a lifecycle commitment. By utilizing the Good Automated Manufacturing Practice (GAMP) framework, we help you align your technical operations with international expectations. This structured methodology allows your team to focus on innovation rather than drowning in redundant paperwork, ensuring that every system from a LIMS to a complex manufacturing execution system remains under control.
The Consequences of Non-Compliance
Ignoring the nuances of validation carries a heavy price. A single 483 observation or Health Canada non-compliance report can stall production for months, damage your market reputation, and lead to significant remediation costs. Beyond the financial impact, poor validation often results in critical data integrity breaches. Since over 60% of regulatory findings now involve computerized systems, these failures directly threaten patient safety and product quality. We help you move from a state of constant anxiety to one of controlled, documented confidence through specialized computer system validation services.
Regulatory Bodies and Global Standards
Achieving global readiness requires a deep understanding of how different agencies interact. While FDA 21 CFR Part 11 simplifies the requirements for electronic records and signatures, Health Canada maintains its own rigorous expectations for system integrity and lifecycle management. The upcoming 2026 EU GMP Annex 11 revision further complicates this by expanding cybersecurity and data integrity requirements to 19 pages of detailed guidance. Aligning your internal protocols with PIC/S and EMA standards ensures your systems remain compliant across international borders. This harmonized approach turns regulatory complexity into a streamlined global strategy that supports long-term business growth.
Implementing a Risk-Based GAMP 5 Framework for GxP Compliance
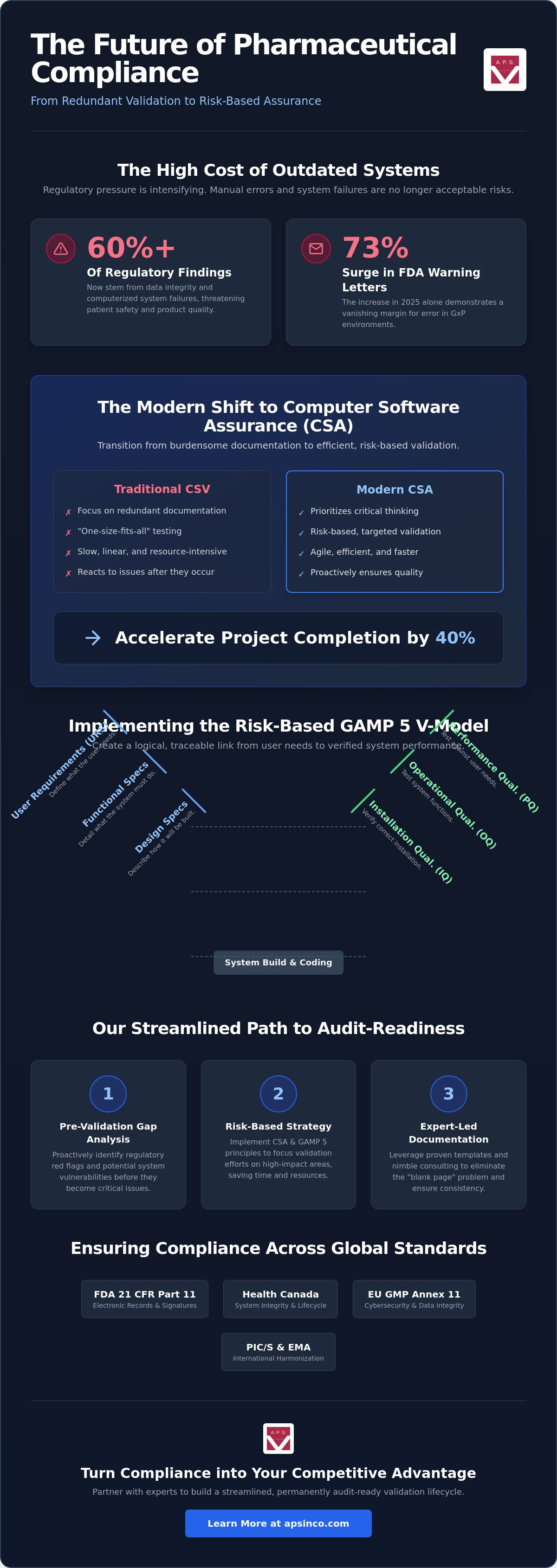
GAMP 5 is the industry gold standard for ensuring computerized systems meet rigorous GxP requirements. It moves beyond simple box-checking to a holistic lifecycle management strategy. By adopting the GAMP 5 framework, organizations align their technical infrastructure with the core principles of quality by design. This approach relies heavily on the V-Model, which creates a direct, traceable link between your User Requirements Specifications (URS) and the final validation testing. It's a method that provides clarity and structure, ensuring every functional requirement is verified before a system goes live.
A "one size fits all" approach fails in modern laboratory environments because the risk profiles of different systems vary wildly. Treating a simple data logger the same as a complex enterprise LIMS leads to wasted resources and documentation bloat. Effective computer system validation services prioritize effort based on the potential impact on patient safety and data reliability. We focus on high-risk areas where failures could compromise product quality, allowing you to streamline the validation of low-risk components. This targeted strategy ensures your compliance efforts are both rigorous and efficient.
Categorising Your Systems (GAMP 5 Categories 1-5)
System categorization is the first step in right-sizing your validation effort. Category 3 covers non-configured software, such as standard off-the-shelf tools where the vendor's own quality assurance provides the primary evidence of compliance. Category 4 involves configured software like PharmaRockIT LIMS, where the system is adapted to fit specific business processes. This requires a more detailed look at configuration specifications and functional testing. Category 5 represents custom-built applications. These pose the highest risk and demand the most comprehensive validation documentation, including deep dives into source code and design specifications.
The Validation Master Plan (VMP)
The Validation Master Plan (VMP) acts as the blueprint for regulatory success. It defines the overall scope, schedule, and responsibilities for site-wide compliance across all computerized systems. A robust VMP integrates Equipment Qualification (IQ/OQ/PQ) into the broader lifecycle, ensuring hardware and software work in perfect harmony. By clearly outlining your validation strategy, you provide auditors with a transparent roadmap of how your facility maintains its GxP status. This level of organization simplifies the audit process and instills confidence in your data integrity protocols.
The Modern Shift: Transitioning from Traditional CSV to Computer Software Assurance (CSA)
The industry is witnessing a pivotal transformation as the FDA moves away from the documentation-heavy Computer System Validation (CSV) model toward a more agile Computer Software Assurance (CSA) approach. This shift, reinforced by the final guidance released in February 2026, addresses a long-standing bottleneck in life sciences; the tendency to prioritize "documentation for documentation’s sake" over actual system performance. By leveraging computer system validation services that embrace CSA, your organization can pivot its focus toward critical thinking and risk-based analysis rather than rote script execution.
CSA doesn't mean less testing. It means smarter testing. Traditional CSV often treats every system with the same level of scrutiny, leading to massive piles of paperwork that don't necessarily improve safety. CSA changes the math. It encourages the use of unscripted testing and vendor-supplied data for lower-risk systems. This modern methodology allows for more exploratory testing, where experts can actually try to break the system rather than just following a pre-written path. It's a more rigorous way to ensure quality while simultaneously removing the bureaucratic weight that often delays digital transformation.
Benefits of Adopting a CSA Approach
Adopting a CSA mindset accelerates your digital evolution. By focusing on the intended use and risk to patient safety, you can reduce documentation burdens by up to 50% for low-risk systems. This significantly lowers the "validation tax" that often prevents laboratories from implementing the latest software. When you streamline these processes, you free up your internal quality teams to focus on high-impact projects, ultimately enhancing the overall systemic integrity of your operations.
When to Stick with Traditional CSV
While CSA is the future, traditional CSV still has its place. High-risk systems that directly interact with product quality or patient safety often require the full weight of scripted testing to satisfy global regulators. During this transition period, a hybrid strategy is often the most pragmatic path. We help you identify which systems are candidates for CSA and which require the traditional rigour of full computer system validation services. This balanced approach ensures you remain compliant with current standards like EU Annex 11 while reaping the efficiency gains of the CSA model.
Streamlining the Validation Lifecycle: From Gap Analysis to Audit Readiness
A well executed validation lifecycle is the difference between a compliant facility and one vulnerable to 483 observations. While the regulatory shift toward CSA discussed in previous sections offers agility, the core execution still requires a disciplined, step by step approach. Professional computer system validation services can accelerate these timelines by 40% by replacing guesswork with proven methodologies. This efficiency begins with the development of robust User Requirement Specifications (URS). A URS isn't just a list of features; it's the foundation of your entire validation project. When written with audit readiness in mind, the URS ensures every functional requirement is measurable, testable, and directly linked to patient safety.
Maintaining a validated state post-implementation requires a lifecycle approach that extends far beyond the initial "go-live" date. It involves continuous monitoring, change control, and periodic reviews to ensure the system doesn't drift out of compliance. By establishing this rigor early, you transform validation from a one-time project hurdle into a sustainable business advantage. This methodical progression ensures that when Health Canada or the FDA arrives for an inspection, your documentation is already complete, organized, and ready for review.
Step 1: Regulatory Gap Analysis and Risk Assessment
Success starts by identifying what you don't know. A comprehensive gap analysis evaluates your legacy systems against modern 21 CFR Part 11 requirements to find hidden "red flags." We prioritize remediation efforts based on the actual risk to your business and patient safety. This step establishes your baseline for audit readiness, ensuring that high risk vulnerabilities are addressed before the formal validation testing begins. It's about being proactive rather than reactive.
Step 2: Execution of IQ, OQ, and PQ
The technical execution phase proves your system is fit for purpose. Installation Qualification (IQ) verifies that the software and hardware are installed correctly according to vendor specifications. Operational Qualification (OQ) pushes the system to its functional limits, testing error handling and security boundaries. Finally, Performance Qualification (PQ) proves the system works reliably within your specific production environment. This triple layered approach provides the documented evidence regulators demand.
Step 3: Final Reporting and Summary
The final phase involves compiling the Validation Summary Report (VSR), which serves as your definitive proof of compliance. This report must demonstrate clear traceability from your initial requirements to the final test results. We ensure all documentation is archived correctly for future inspections, leaving no room for ambiguity. If you're ready to accelerate your path to compliance, explore our computer system validation services to see how we can streamline your next project.
Maximising ROI with Expert Validation Consulting and Integrated Software Solutions
Choosing a partner for computer system validation services is a strategic decision that impacts your bottom line as much as your compliance status. While massive, impersonal firms often apply rigid, one-size-fits-all models, a nimble and expert-led partner like APS Compliance Consultants Inc. prioritizes efficiency and direct relationships. We understand that time is your most valuable asset. By utilizing our library of industry-vetted templates, you bypass the "blank page" hurdle that often stalls documentation for weeks. This allows your team to focus on their primary roles while we handle the technical rigour of GxP adherence.
The APS Advantage: Speed and Precision
Our team accelerates projects by 40% using refined methodologies that have been tested in the most demanding regulatory environments. Being based in North America, we offer specialized expertise for Health Canada and FDA projects, ensuring your systems meet local and international expectations simultaneously. We view our work as a collaborative partnership rather than a transactional service. This means we're invested in your long-term success, not just the next milestone. You gain access to consultants who are equally comfortable discussing technical standards and practical business growth.
To further capitalize on your operational efficiencies and drive growth, you can explore AI lead Gen to automate your sales pipeline with specialized digital representatives from Global AI Reps.
Sustaining Compliance with Integrated SaaS
The future of life sciences lies in cloud-based, pre-validated software. By using Alleye CMMS for asset management and PharmaRockIT for lab management, you're investing in systems built for modern, audit-ready environments. These solutions reduce the ongoing validation burden, allowing your facility to remain agile as regulations evolve. This systemic approach to integrity means you don't just achieve compliance; you sustain it with minimal effort, even as your operations scale.
Next Steps for Your Validation Project
Every system is unique, and your validation strategy should reflect that. Whether you need a comprehensive regulatory gap analysis or a targeted system assessment, we're here to help you find the most efficient path forward. We can customize a strategy that fits your specific budget and timeline without compromising on quality or safety. To begin your journey toward seamless compliance, schedule a consultation with APS Compliance Consultants Inc. today.
Future-Proofing Your Compliance Strategy
The regulatory landscape of 2026 leaves no room for stagnant validation practices. As we've explored, the transition toward Computer Software Assurance and the risk-based rigour of GAMP 5 are not merely trends; they're survival strategies for the modern life sciences industry. Moving beyond the "documentation for documentation’s sake" mindset allows your organization to prioritize actual system performance and patient safety. This evolution transforms compliance from a reactive burden into a proactive shield against the complexities of global inspections.
Sustainable success depends on a disciplined lifecycle approach that integrates your daily workflows with robust data integrity controls. By leveraging specialized computer system validation services, you ensure that every update, configuration, and system retirement is handled with the precision required by Health Canada and the FDA. This strategic alignment creates a culture of quality that persists long after the initial qualification is complete, ensuring your operations remain agile and resilient in the face of evolving standards.
Accelerate your compliance project with APS Compliance Consultants Inc. expert CSV services to secure your facility's future. Our team is ready to help you navigate these high-stakes requirements with authority and a collaborative spirit. Let's build a more reliable, audit-ready environment together.
Frequently Asked Questions
What are computer system validation services?
These are professional consulting activities that provide documented evidence that a computerized system consistently performs its intended function in a GxP environment. These services cover the entire lifecycle, from initial risk assessment and User Requirement Specifications to the final Validation Summary Report. By partnering with experts, you ensure that critical software like PharmaRockIT LIMS or Alleye CMMS remains compliant with global regulatory standards.
How long does a typical CSV project take to complete?
Timelines vary based on system complexity, but a standard validation project typically ranges from four to twelve weeks. Simple off-the-shelf software might be validated quickly, while custom-built applications requiring deep technical testing take longer. We focus on streamlining these phases through pre-validation gap analyses, which identify potential roadblocks before they cause delays in your production schedule.
Is GAMP 5 a regulatory requirement for Health Canada?
GAMP 5 is not a legal requirement, but it is the industry-standard methodology that Health Canada inspectors expect to see used. It provides a structured, risk-based framework that demonstrates your facility is following current Good Manufacturing Practices (cGMP). Aligning your processes with this guide ensures your documentation meets the rigorous expectations for data integrity and system reliability during an official inspection.
What is the difference between CSV and CSA?
The primary difference lies in the shift from documentation-heavy protocols to a focus on critical thinking and risk-based testing. Traditional CSV often requires extensive scripted testing for every feature, whereas Computer Software Assurance (CSA) encourages unscripted testing and vendor-supplied data for low-risk functions. This modern approach reduces the documentation burden significantly while maintaining high standards for patient safety and product quality.
Can I use Excel spreadsheets for GxP data if they are validated?
Yes, you can use Excel for GxP data, provided the spreadsheets are fully validated and protected against unauthorized changes. This involves locking cells with formulas, implementing version control, and ensuring an audit trail for data entry. Many organizations are moving away from spreadsheets toward more robust, pre-validated solutions like PharmaRockIT LIMS to reduce the risk of human error and data integrity breaches.
How often do computerized systems need to be revalidated?
Systems should be revalidated whenever a significant change occurs, such as a software upgrade, hardware replacement, or a change in intended use. Additionally, periodic reviews should be conducted every one to two years to confirm the system remains in a validated state. This proactive approach ensures that your operations stay compliant with 21 CFR Part 11 and other global standards throughout the system's lifecycle.
What are the main 21 CFR Part 11 requirements for software?
The core requirements focus on ensuring the authenticity, integrity, and confidentiality of electronic records and signatures. This includes maintaining secure, computer-generated audit trails, implementing strict access controls, and using digital signatures that are uniquely linked to the signer. Our computer system validation services specialize in verifying that your technical controls and procedural safeguards meet these specific FDA mandates for electronic data.
How does APS Compliance Consultants Inc. accelerate the validation process?
We accelerate project timelines by up to 40% by utilizing a library of proven, industry-vetted templates and a risk-based methodology. Instead of starting from scratch, we leverage our deep expertise in GAMP 5 and Health Canada standards to bypass common documentation hurdles. This agile approach allows your team to focus on core operations while we handle the technical rigour of achieving audit readiness.



Comments